Software
Analytical tools for LC-MS/MS
DeepMASS2
DeepMASS2 is a cross-platform GUI software tool, which enables deep-learning based metabolite annotation via semantic similarity analysis of mass spectral language. This approach enables the prediction of structurally related metabolites for the unknown compounds. By considering the chemical space, these structurally related metabolites provide valuable information about the potential location of the unknown metabolites and assist in ranking candidates obtained from molecular structure databases.
Web Link:
Paper:
- Under Review
Analytical tools for GC-EI/MS
FederEI
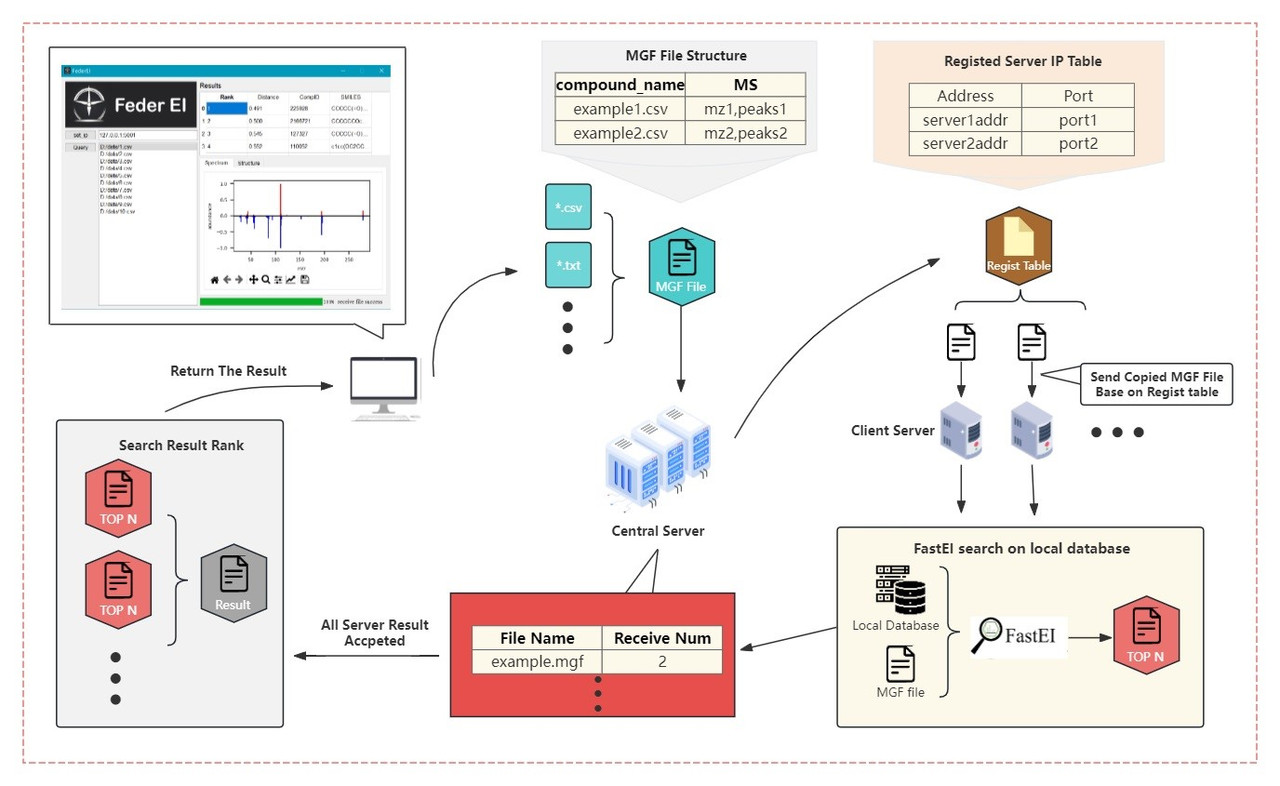
FederEI is a federated library matching solution for EI-MS-based compound identification. It establishes a server-to-server connection framework seamlessly integrated into a user-friendly front-end software. By keeping data localized within each laboratory's server, FederEI minimizes the need for sharing sensitive spectral information across multiple entities, thus mitigating privacy concerns.
Here is the workflow: The user submits mass spectrometry data through the user interface (UI), and FederEI dispatches it to the central-server. Upon reception, the central-server distributes the file to all client-server(s) based on their respective IP addresses stored in the client-server registry. Subsequently, each client-server searches for results in its local database using library matching algorithm and transmits them back to the central-server. The central-server tallies the received search results for the file until all client-servers have responded. Finally, central-server organizes and summarizes the results, and return the final results to the corresponding user.
Web Link:
Paper:
- Under Review
Analytical tools for thermal proteomics
ProSAP
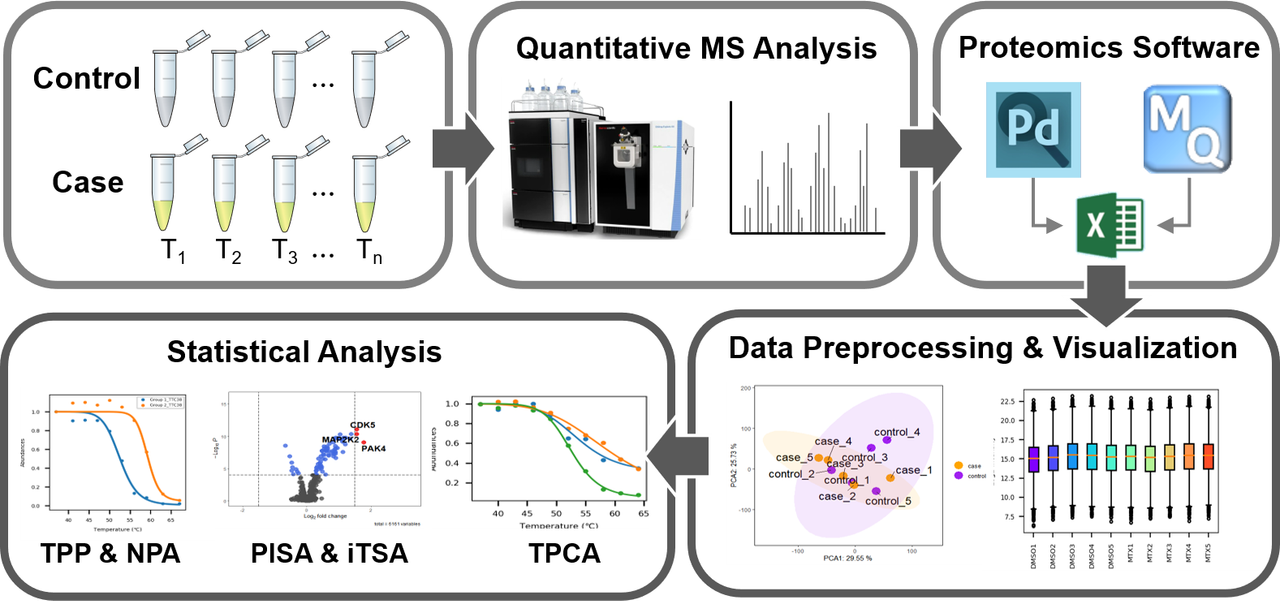
ProSAP (Protein Stability Analysis Pod) is standalone and user-friendly software with graphical user interface (GUI). ProSAP provides an integrated analysis workflow for thermal shift assay, which includes five modules: data preprocessing, data visualization, TPP analysis, NPARC analysis and iTSA analysis. With the assistance of the user-friendly interface, researchers can easily compare several statistical strategies, analyze the results and draw the conclusion from the proteomics quantitative table obtained by Proteome Discoverer or MaxQuant. Users would also benefit from a comprehensive overview of the performance of different algorithms, and apply appropriate algorithms to their dataset easily.
Web Link:
Paper:
- Ji, H.; Lu, X.; Zheng, Z.; Sun, S.; Tan, C.S.H. ProSAP: A GUI Software Tool for Statistical Analysis and Assessment of Thermal Stability Data. Brief. Bioinform. 2022, 23 (3), bbac057. link
MAPS-iTSA
Target deconvolution is a crucial but costly and time-consuming task that hinders large-scale profiling for drug discovery. We present a matrix-augmented pooling strategy (MAPS) which mixes multiple drugs into samples with optimized permutation and delineates targets of each drug simultaneously with mathematical processing. We validated this strategy with thermal proteome profiling (TPP) testing of 15 drugs concurrently, increasing experimental throughput by 60x while maintaining high sensitivity and specificity. Benefiting from the lower cost and higher throughput of MAPS, we performed target deconvolution of the 15 drugs across 5 cell lines.
Web Link:

Paper:
- Ji, H.#; Lu, X.#; Zhao, S.; Wang, Q.; Bin, L.; Huber, K. V. M.; Luo, R.; Tian, R.; Tan, C. S. H. Target deconvolution with matrix-augmented pooling strategy reveals cell-specific drug-protein interactions. Cell Chem. Biol. 2023, 30(11) 1478-1487. link
Other bioinformatics software
PyFingerprint
There are many types of chemical fingerprint for describing the molecule provided by different tools, such as RDKit, CDK and OpenBabel. This package aims to summarize them all in PyFingerprint.
Web Link: